Part1. introduction
1. Overview
The Mixed Effect Neural Networks (NN-MM) extend linear mixed model ("MM") to multilayer neural networks ("NN") by adding one middle layer between genotype layer and phenotypes layer. Nodes in the middle layer represent intermediate traits, e.g., the known intermediate omics features such as gene expression levels can be incorporated in the middle layer. These three sequential layers form a unified network.

NN-MM allow any patterns of missing data in the middle layer, and missing data will be sampled. In below figure, for an individual, the gene expression levels of the first two genes are 0.9 and 0.1, respectively, and the gene expression level of the last gene is missing to be sampled. The missing patterns of gene expression levels can be different for different individuals.
2. Extend linear mixed model to multilayer neural networks
Multiple independent single-trait mixed models are used to model the relationships between input layer (genotypes) and middle layer (intermediate traits). Activation functions in the neural network are used to approximate the linear/nonlinear relationships between middle layer (intermediate traits) and output layer (phenotypes). Missing values in the middle layer (intermediate traits) are sampled by Hamiltonian Monte Carlo based on the upstream genotype layer and downstream phenotype layer.
Details can be found in our publications:
- Tianjing Zhao, Jian Zeng, and Hao Cheng. Extend mixed models to multilayer neural networks for genomic prediction including intermediate omics data, GENETICS, 2022; https://doi.org/10.1093/genetics/iyac034.
- Tianjing Zhao, Rohan Fernando, and Hao Cheng. Interpretable artificial neural networks incorporating Bayesian alphabet models for genome-wide prediction and association studies, G3 Genes|Genomes|Genetics, 2021; https://doi.org/10.1093/g3journal/jkab228
3. Flexibility
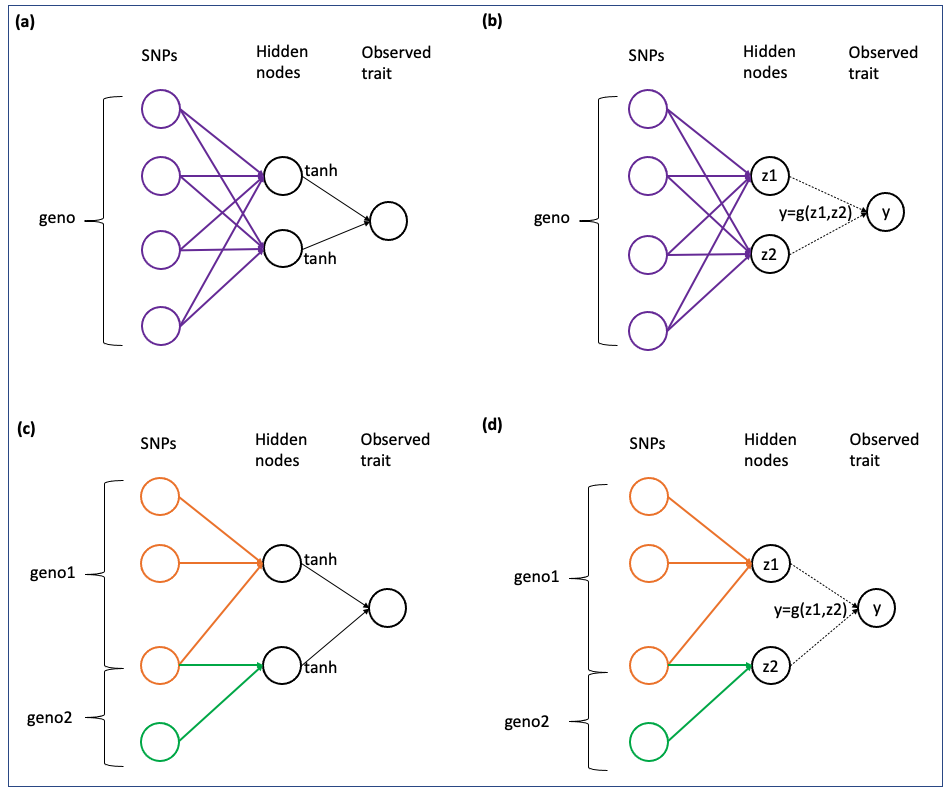
NN-MM can fit fully-connected neural networks ((a),(b)), or partial-connected neural networks ((c),(d)). Also, the relationship between middle layer (intermediate traits) and output layer (phenotypes) can be based on activation functions ((a),(c)), or pre-defined by a user-defined function ((b),(d)).
4. Multi-threaded parallelism
By default, multiple single-trait models will be used to model the relationships between input layer (genotypes) and middle layer (intermediate traits). Multi-threaded parallelism will be used for parallel computing. The number of threads can be checked by running Threads.nthreads() in Julia. Usually, using multiple threads will be about 3 times faster than using a single thread.
The number of execution threads is controlled by using the -t/–threads command-line argument (requires at least Julia 1.5).
For example, to start Julia with 4 threads:
julia --threads 4If you're using Juno via IDE like Atom, all threads will be loaded automatically.