Part 3: NNMM with Intermediate Omics Features
This documentation reflects NNMM.jl v0.3+ using the Layer/Equation/runNNMM API.
Model Architecture
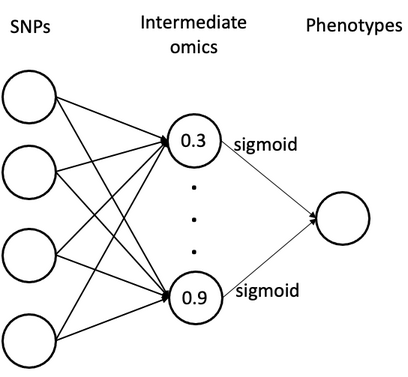
When you have observed intermediate omics data (e.g., gene expression, metabolomics), NNMM can incorporate them in the middle layer:
Genotypes → (Complete/Incomplete) Intermediate Omics → Phenotypes
(Layer 1) (Layer 2) (Layer 3)- Omics feature names are specified in the
omics_nameparameter of the firstEquation - Missing omics values are allowed and will be sampled via HMC
- Testing individuals (without phenotypes) can still contribute omics data to improve the genotype→omics model
Example: Fully-Connected Network with Observed Omics
This example demonstrates:
- Activation function:
sigmoid(other options:"tanh","relu","leakyrelu","linear") - Number of omics features: 10
- Bayesian model: BayesC for marker effects
- Missing omics handling: Hamiltonian Monte Carlo
# Step 1: Load packages
using NNMM
using NNMM.Datasets
using DataFrames
using CSV
using Statistics
using Random
Random.seed!(123)
# Step 2: Load simulated dataset
geno_path = Datasets.dataset("genotypes_1000snps.txt", dataset_name="simulated_omics_data")
pheno_path = Datasets.dataset("phenotypes_sim.txt", dataset_name="simulated_omics_data")
# Read data
pheno_df = CSV.read(pheno_path, DataFrame)
# Step 3: Prepare omics file (Layer 2)
# Select ID and 10 omics features
omics_cols = vcat(:ID, [Symbol("omic$i") for i in 1:10])
omics_df = pheno_df[:, omics_cols]
omics_names = names(omics_df)[2:end] # ["omic1", "omic2", ..., "omic10"]
omics_file = "omics_data.csv"
CSV.write(omics_file, omics_df; missingstring="NA")
# Step 4: Prepare phenotype file (Layer 3)
trait_df = pheno_df[:, [:ID, :trait1]]
trait_file = "phenotypes_data.csv"
CSV.write(trait_file, trait_df; missingstring="NA")
# Step 5: Define layers
layers = [
# Layer 1: Genotypes
Layer(
layer_name = "genotypes",
data_path = [geno_path]
),
# Layer 2: Omics (with observed values)
Layer(
layer_name = "omics",
data_path = omics_file,
missing_value = "NA"
),
# Layer 3: Phenotypes
Layer(
layer_name = "phenotypes",
data_path = trait_file,
missing_value = "NA"
)
]
# Step 6: Define equations
equations = [
# Genotypes → Omics (BayesC)
Equation(
from_layer_name = "genotypes",
to_layer_name = "omics",
equation = "omics = intercept + genotypes",
omics_name = omics_names, # Names of omics columns
method = "BayesC",
estimatePi = true
),
# Omics → Phenotypes (sigmoid activation)
Equation(
from_layer_name = "omics",
to_layer_name = "phenotypes",
equation = "phenotypes = intercept + omics",
phenotype_name = ["trait1"],
method = "BayesC",
activation_function = "sigmoid"
)
]
# Step 7: Run analysis
out = runNNMM(layers, equations;
chain_length = 5000,
burnin = 1000,
printout_frequency = 2000, # Print every 2000 iterations
output_folder = "nnmm_omics_results"
)
# Step 8: Check accuracy
ebv = out["EBV_NonLinear"]
# Convert ID types for joining (EBV IDs may be of type Any)
ebv.ID = string.(ebv.ID)
pheno_df.ID = string.(pheno_df.ID)
# If true breeding values available:
if hasproperty(pheno_df, :genetic_total)
results = innerjoin(ebv, pheno_df[:, [:ID, :genetic_total]], on=:ID)
accuracy = cor(Float64.(results.EBV), results.genetic_total)
println("Prediction accuracy: ", round(accuracy, digits=4))
end
# Cleanup
rm(omics_file, force=true)
rm(trait_file, force=true)Including Residual Polygenic Effects
To include a residual polygenic component (genetic effects not mediated by omics), add an extra latent node to the middle layer:
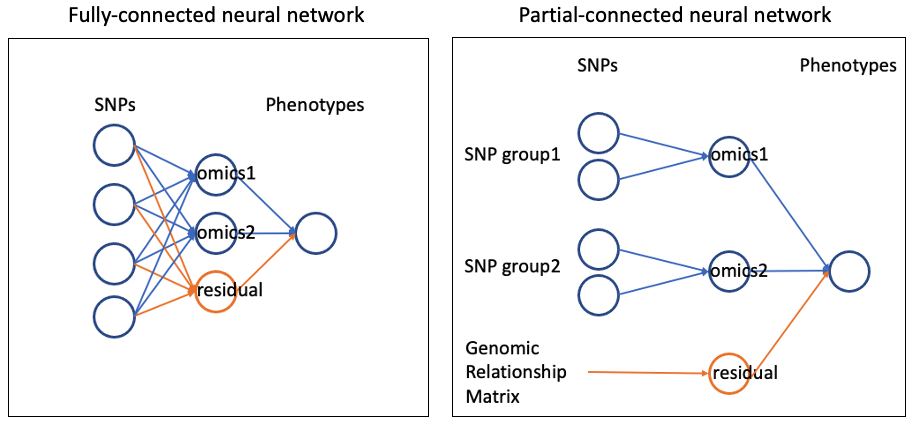
Example: Network with Residual Polygenic Effect
# Step 1: Load packages
using NNMM
using NNMM.Datasets
using DataFrames
using CSV
using Statistics
using Random
Random.seed!(123)
# Step 2: Load data
geno_path = Datasets.dataset("genotypes_1000snps.txt", dataset_name="simulated_omics_data")
pheno_path = Datasets.dataset("phenotypes_sim.txt", dataset_name="simulated_omics_data")
pheno_df = CSV.read(pheno_path, DataFrame)
# Step 3: Create omics file WITH an extra "residual" column (all missing)
omics_cols = vcat(:ID, [Symbol("omic$i") for i in 1:10])
omics_df = pheno_df[:, omics_cols]
# Add residual column (all missing - will be sampled)
omics_df[!, :residual] = fill(missing, nrow(omics_df))
omics_names = names(omics_df)[2:end] # ["omic1", ..., "omic10", "residual"]
omics_file = "omics_with_residual.csv"
CSV.write(omics_file, omics_df; missingstring="NA")
# Step 4: Create phenotype file
trait_df = pheno_df[:, [:ID, :trait1]]
trait_file = "phenotypes_data.csv"
CSV.write(trait_file, trait_df; missingstring="NA")
# Step 5: Define layers
layers = [
Layer(layer_name = "genotypes", data_path = [geno_path]),
Layer(layer_name = "omics", data_path = omics_file, missing_value = "NA"),
Layer(layer_name = "phenotypes", data_path = trait_file, missing_value = "NA")
]
# Step 6: Define equations (11 features: 10 omics + 1 residual)
equations = [
Equation(
from_layer_name = "genotypes",
to_layer_name = "omics",
equation = "omics = intercept + genotypes",
omics_name = omics_names, # Includes "residual"
method = "BayesC",
estimatePi = true
),
Equation(
from_layer_name = "omics",
to_layer_name = "phenotypes",
equation = "phenotypes = intercept + omics",
phenotype_name = ["trait1"],
method = "BayesC",
activation_function = "sigmoid"
)
]
# Step 7: Run analysis
out = runNNMM(layers, equations;
chain_length = 5000,
burnin = 1000,
output_folder = "nnmm_residual_results"
)
# Step 8: Check accuracy
ebv = out["EBV_NonLinear"]
# Convert ID types for joining
ebv.ID = string.(ebv.ID)
pheno_df.ID = string.(pheno_df.ID)
if hasproperty(pheno_df, :genetic_total)
results = innerjoin(ebv, pheno_df[:, [:ID, :genetic_total]], on=:ID)
accuracy = cor(Float64.(results.EBV), results.genetic_total)
println("Prediction accuracy: ", round(accuracy, digits=4))
end
# Cleanup
rm(omics_file, force=true)
rm(trait_file, force=true)Handling Missing Omics Data
NNMM automatically handles missing omics values in the training set. Missing values are sampled using HMC based on:
- The upstream genotype layer (marker effects)
- The downstream phenotype layer (via backpropagation)
Setting Missing Values Manually
# Convert column type to allow missing values
omics_df[!, :omic1] = convert(Vector{Union{Missing, Float64}}, omics_df[!, :omic1])
# Set specific values to missing
omics_df[10:15, :omic1] .= missing # Set rows 10-15 as missing for omic1
# Set phenotypes for testing individuals as missing
pheno_df[!, :trait1] = convert(Vector{Union{Missing, Float64}}, pheno_df[!, :trait1])
pheno_df[90:100, :trait1] .= missing # Testing individualsOutput Files
The output files are the same as described in Part 2, with omics names replacing latent node names.
Tips
Many omics features: For large numbers of omics (>100), set
printout_frequencyto a large value to reduce console output.Pre-processing: Center and scale omics data before running NNMM for better convergence.
Testing individuals: Include individuals without phenotypes if they have omics data - this improves the genotype→omics model.
Residual effects: Add a completely missing "residual" column to capture genetic variance not explained by omics.